![[Prev]](p_prev.gif)
![[Next]](p_next.gif)
Die Regeln, die für die in 6.10.2 gezeigten simulierten Massenspektren verwendet wurden, basieren auf den gleichen neuronalen Netzen für die Bewertung, die auch mit MASSIMO eingesetzt wurden (siehe 4.2.5.2). Für die Berechnung der Konzentrationen wird, wie in MASSIMO, die gleiche Wahrscheinlichkeitskinetik verwendet, die sich allerdings bei der Behandlung der Umlagerungen unterscheidet. Wie in Kapitel 4.3.3.1 gesagt, gilt die Annahme nicht ganz, daß sich die Wasserstoffumlagerungen im Gleichgewicht befinden, die von MASSIMO für die Konzentrationsberechnungen für die Wasserstoffumlagerung eingesetzt wird. Deshalb wird für diese Reaktionen bei EROS7 statt der Gleichgewichtskonstante der Prozentsatz angegeben, der bei einer Symmetriezahl von eins von dieser Umlagerung umgesetzt wird. EROS7 berücksichtigt dabei auch, wenn das Produkt wieder zurücklagern kann und eine entsprechende Reaktion durchgeführt wurde.
Die Regeln enthalten sechs Reaktionstypen: die Ionisation von freien Elektronenpaaren und 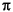 -Systemen, denen kein freies Elektronenpaar benachbart ist (in Anlehnung an die Reaktionsgenerierung mit MASSIMO), die
-Systemen, denen kein freies Elektronenpaar benachbart ist (in Anlehnung an die Reaktionsgenerierung mit MASSIMO), die  -Spaltung,
die Oniumreaktion, die Carbonyleliminierung, die McLafferty-Spaltung und die Wasserstoffumlagerung. Die Bewertungen wurden ebenso aus den Regeln von MASSIMO übernommen, wie die Reaktionstypen. Die Ionisation und die Wasserstoffumlagerung konnten nicht
direkt übertragen werden und wurden deshalb an die Version von MASSIMO angelehnt. Ohne diese Anlehnung hätte man nicht mit einer sinnvollen Bewertung der Reaktionen rechnen können. Eine Besonderheit stellt dabei die Ionisation dar. Sie ist unabhängig
von jeglichem Atom und hat damit kein Atom in der Definition der Reaktionssubstruktur. Es gibt dennoch mehrere verschiedene Ionisationsmöglichkeiten in einem Molekül, nämlich so viele, wie es unterschiedliche, besetzte Elektronensysteme im Molekül
gibt. Die Ionisation sucht diese, unter der oben angegebenen Beschränkung und führt sie durch. Für jede Reaktion wird zunächst das Ensemble eins, das Edukt, als aktives Ensemble gesetzt und von ihm ausgehend alle Reaktionen durchgeführt.
-Spaltung,
die Oniumreaktion, die Carbonyleliminierung, die McLafferty-Spaltung und die Wasserstoffumlagerung. Die Bewertungen wurden ebenso aus den Regeln von MASSIMO übernommen, wie die Reaktionstypen. Die Ionisation und die Wasserstoffumlagerung konnten nicht
direkt übertragen werden und wurden deshalb an die Version von MASSIMO angelehnt. Ohne diese Anlehnung hätte man nicht mit einer sinnvollen Bewertung der Reaktionen rechnen können. Eine Besonderheit stellt dabei die Ionisation dar. Sie ist unabhängig
von jeglichem Atom und hat damit kein Atom in der Definition der Reaktionssubstruktur. Es gibt dennoch mehrere verschiedene Ionisationsmöglichkeiten in einem Molekül, nämlich so viele, wie es unterschiedliche, besetzte Elektronensysteme im Molekül
gibt. Die Ionisation sucht diese, unter der oben angegebenen Beschränkung und führt sie durch. Für jede Reaktion wird zunächst das Ensemble eins, das Edukt, als aktives Ensemble gesetzt und von ihm ausgehend alle Reaktionen durchgeführt.
An den verwendeten Regeln zur Simulation von Massenspektren, die im Anhang F.2 gezeigt sind, sieht man neben der Variablenübergabe von EROS7 an die Regeln (Reaktionsebene) und der Erzeugung mehrerer Reaktionen aus einer
gefundenen Reaktionssubstruktur (Ionisationsregel) weitere Möglichkeiten der Programmierung der Regeln. So wird das radikalische Elektronensystem nur einmal für jedes Edukt gesucht und seine Nummer in einer statischen Variablen gespeichert. Bei der
Überprüfung der Bedingungen am Reaktionszentrum und während der Reaktion selbst wird dann auf diese zugegriffen. Desweiteren setzen die Regeln Backpropagationnetzwerke zur Ermittlung der Reaktionswahrscheinlichkeit ein. Für das Edukt und
das Produkt werden die Funktionen zum Setzen der Eingabewerte des Netzes aufgerufen, um schließlich beim Abholen des Ergebnisses das neuronale Netz auszuwerten. Hat man aufwendigere, klar umrissene Aufgaben, die für mehrere Reaktionstypen benötigt
werden, oder, um die Regeln leichter lesen zu können, kann man sich für diese Aufgaben eigene Funktionen dafür schreiben. Dies geschah für die Dekonjugation von -Systemen ohne richtigen Bindungsbruch, die Zählung
von freien Elektronenpaaren an Atomen, die an einem bestimmten -System beteiligt sind, die Abfrage von ganzzahligen Eigenschaften, die als Funktionswert zurückgegeben werden und damit manche Stelle der Regeln übersichtlicher
gestalten, und die Berechnung des Ionisationspotentials. Die Berechnung des Ionisationspotentials nach [26][46], die in MASSIMO eingebaut war, ist nicht so leicht an die Ionisation
aus Elektronensystemen anzupassen, weshalb die Funktion in den Regeln immer den Wert 10.0 eV zurückgibt. Mit dem Vorfaktor und der Kennzeichnung als Umlagerungsreaktion werden die Ionisationsprodukte proportional zur Zahl gleicher Elektronensysteme gebildet.
Diese Regeln können also nur für Verbindungen angewendet werden, deren verschiedene Ionisationsorte sehr ähnliche Ionisationspotentiale besitzen. Überdies sind die Regeln so gestaltet, daß man über eine Option steuern kann, ob
Wasserstoffumlagerungen stattfinden sollen oder nicht. Zur Aktivierung wird EROS7 mit dem entsprechenden Aufrufparameter mitgeteilt, daß es eine Variable vom Typ int unter dem Namen huml und dem Wert 1 an die Regeln übergeben soll. Wird diese Option
beim Aufruf von EROS7 weggelassen, werden keine Wasserstoffumlagerungen durchgeführt, da dies in den Regeln so definiert ist.
Die Weise, in der EROS7 das Reaktionsnetzwerk aufbaut, unterscheidet sich von der von MASSIMO speziell für die Umlagerungen. MASSIMO hat diese für jedes Umlagerungssystem im ganzen vor den Fragmentierungen durchgeführt und bewertet. In EROS7 wird dies alles von der Kinetik übernommen und auch in Fällen, bei denen ein Reaktionsprodukt erneut über eine andere Reaktionssequenz gebildet wird, werden von EROS7 im Gegensatz zu MASSIOMO die Reaktionen, die von diesem Kation ausgehen, nicht erneut generiert.
Da die Datenstruktur RICOS auch die Isotopenmarkierung von einzelnen Atomen kennt, ist es mit EROS7 möglich, für solche Verbindungen das Massenspektrum zu berechnen.
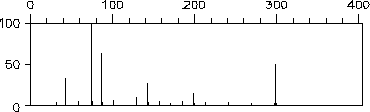
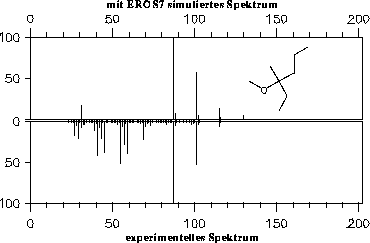
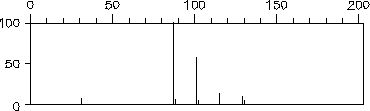
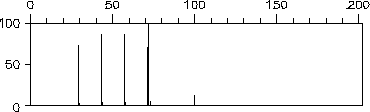
Die simulierten Massenspektren von (1-Ethyl-1-methyl-butyl)-methyl-ether in Abbildung 131 und 3-Hexanon in Abbildung 133 wurden unter Ausschluß der Wasserstoffumlagerung berechnet. Die Absenkung der Abspaltung von Wasserstoffradikalen, wie sie bei der Verwendung der Wasserstoffumlagerung benötigt wird, war jedoch in den Regeln enthalten. Zum Vergleich zeigen Abbildung 132 und Abbildung 134 die von MASSIMO simulierten Spektren.
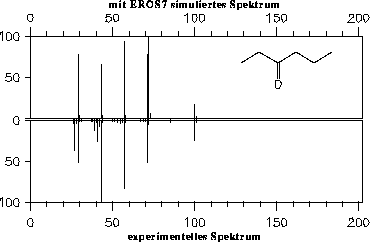
 43 und 5729) mit 0.454
und 0.453 auch fast gleich sind und auch die Symmetriezahlen in beiden Fällen eins sind, entspricht das Spektrum von EROS7 den Wahrscheinlichkeiten, obwohl das nicht ganz korrekt berechnete Spektrum von MASSIMO näher am experimentellen Massenspektrum
liegt.
43 und 5729) mit 0.454
und 0.453 auch fast gleich sind und auch die Symmetriezahlen in beiden Fällen eins sind, entspricht das Spektrum von EROS7 den Wahrscheinlichkeiten, obwohl das nicht ganz korrekt berechnete Spektrum von MASSIMO näher am experimentellen Massenspektrum
liegt.Für die Wasserstoffumlagerungen wurden wegen der geänderten Kinetik für die Umlagerungen neue Reaktivitäten bestimmt. Abbildung 135 zeigt den Spektrenvergleich: simuliertes gegen das experimentelle Massenspektrum von Methylstearat. Zum Vergleich ist das mit MASSIMO simulierte Spektrum von Methylstearat in Abbildung 136 dargestellt.