![[Prev]](p_prev.gif)
![[Next]](p_next.gif)
proc Tcl_rule {} {
global irule op sub_op rule_trace
global_name
global rname rdate kin_mode convlim rxtime minconc k radic_els rctr level
global phases_p_r phase_contacts phprop comp_edus in_phase ophase
global attrib_1 center_1 rule_1
...
switch $irule {
GLOBAL {
switch $op {
INIT_RULES {
set rname mix-ms.tcl"
putco name rname
set rdate 30.09.1997"
putco date rdate
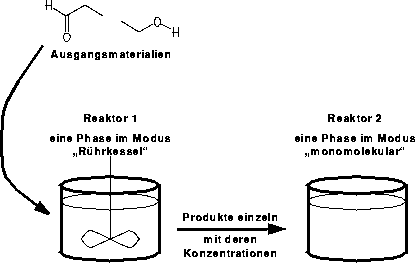
# Reactoren und Phasen
# 2 Reaktoren mit je einer Phase
set phases_p_r {1,1,0}
# Phasen mit den Ausgangsmaterialien fuer die Reaktoren
# Reaktor 1: aus Datei, Reaktor 2: Phase 1
set in_phase {0,1}
# in den Reaktor 1 kommen alle Edukte zusammen,
# in den Reaktor 2 getrennt nacheinander
set comp_edus {1,0}
# keine Phasenkontakte
set phase_contacts(0) 0
# Phasenmodi
set phprop(0) $MIX"
set phprop(1) $MONOMOLEC"
# Definieren der Ausgabephase
set ophase 2
# Kinetikmodi der Reaktoren
set kin_mode(0) gear
set kin_mode(1) prob_kin
# Initialisieren der Reaktivitaetsvariablen
set k 0.0
# min. Konzentration, Reaktionszeit und geforderter Umsatz
set minconc 1.0e-5
set rxtime 3600.
set convlim 0.95
# Uebergbe der Variablen an EROS7
putco phases_per_reactor phases_p_r
putco phase_contacts phase_contacts
putco phase_property phprop
putco use_all_educts_together comp_edus
putco input_phase_for_reactors in_phase
putco output_phase ophase
putco kinetic_model kin_mode
put reactivity k
putco minimal_concentration minconc
putco reaction_time rxtime
putco conversion_limit convlim
# Holen der Variablen reactor und level von EROS7
get reactor rctr
get level level
return OK
}
...
In den beiden Reaktoren laufen überdies nicht nur Reaktionen mit verschiedenem Typ ab, sondern liefern auch einmal eine Geschwindigkeitskonstante und im anderen Fall eine Reaktionswahrscheinlichkeit. Die Regeln enthalten alle möglichen Reaktionstypen
und überprüfen mit den Bedingungen des gesamten Eduktensembles (CONSTR, sub_op=0) auch, ob dieser Reaktionstyp im aktuellen Reaktor stattfindet.
...
CONSTR {
switch $sub_op {
0 {
if {$rctr != 1} {return BAD}
...
return OK
}
...
Die Ionisation ist beispielweise nur im Reaktor zwei zugelassen. Um dies entscheiden zu können, werden von EROS7 während der Initialisierung der Regeln Variablen übergeben, die immer den aktuellen Reaktor, die Phase und die Reaktionsebene enthalten.
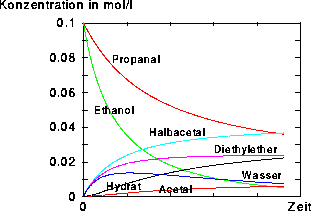
Hat man sich bei der Regelinitialisierung den Zeiger auf die Variable mit der Reaktornummer abgeholt, kann mit den Bedingungen des Eduktensembles auch festgelegt werden, welche Reaktionstypen im aktuellen Reaktor stattfinden. Für den Reaktor eins waren Reaktionstypen zugelassen, die aus Propanal und Ethanol säurekatalysiert das Halbacetal, Acetal und den Diethylether, sowie zusammen mit dem dabei entstehenden Wasser auch das Hydrat des Propanals gebildet haben. Zur Simulation der Massenspektren im Reaktor zwei wurden die Ionisation und Fragmentierungen eingesetzt, die auch schon zur ausschließlichen Massenspektrenvorhersage (siehe Kapitel 6.10.2 und 7.2) verwendet wurden. Auf die Wasserstoffumlagerung wurde dabei verzichtet.
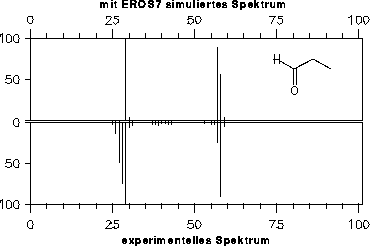
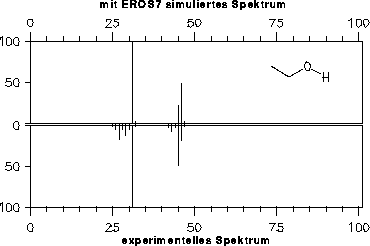
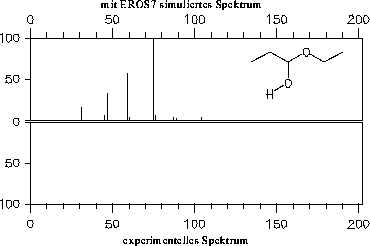
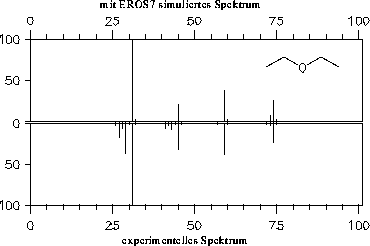
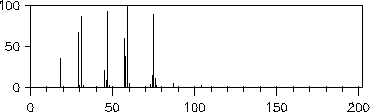
Da die Signale des Massenspektrums vor der Ausgabe von EROS7 so skaliert werden, daß der Basepeak 100% erhält, hat die vom Reaktor eins bestimmte Endkonzentration der Substanz keinen Einfluß auf die Spektren, weshalb hier nicht darauf eingegangen wird (im Gegensatz zum nächsten Kapitel 7.3.2).
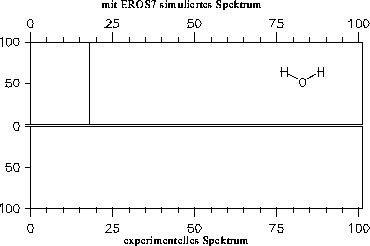
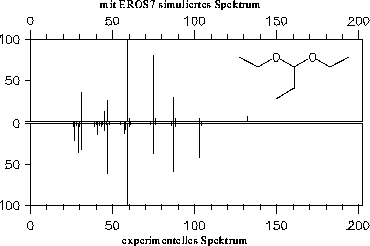
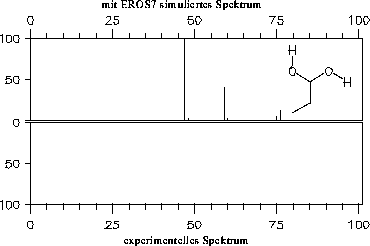
Die simulierten Massenspektren sind zusammen mit einem experimentellen Spektrum zum Vergleich in den Abbildungen 202 bis 208 dargestellt. Für drei der Verbindungen stand allerdings kein experimentelles Massenspektrum zur Verfügung: Halbacetal, Wasser und Propanalhydrat. Vom Propanalhydrat und Halbacetal dürfte die Stabilität der Verbindungen nicht ausreichen, daß von ihnen selbst ein Massenspektrum aufgenommen werden kann. Sie werden schon vor der Ionisation Wasser bzw. Ethanol abspalten. Ein solches Verhalten wird jedoch nicht von den Fragmentierungsregeln berücksichtigt. Statt der angegebenen, simulierten Spektren müßte das Gemischspektrum der Spaltungsprodukte angegeben werden.