![[Prev]](p_prev.gif)
![[Next]](p_next.gif)
bestimmen. Eine Beschränkung der Zahl der Zwischenprodukte und Übergangszustände gibt es nicht. Zur schnelleren Abarbeitung der Regeln von EROS7 sollten nach Möglichkeit alle Modifikationen in einem Block durchgeführt werden und zwischendurch keine Eigenschaften abgefragte werden, die auch vom Edukt abgefragt und zwischengespeichert werden können, damit möglichst wenig Zwischenprodukte erzeugt werden.
Von den Edukten über die Zwischenprodukte und Übergangszustände zu den Produkten muß gewährleistet sein, daß jedes Atom nach den einzelnen Reaktionsteilschritten auch wiedergefunden werden kann. Deshalb wird gewährleistet, daß der Index für alle Atome, Elektronensysteme und Gruppen während aller Teilreaktionen gleich bleibt. Während der Reaktionen können allerdings Elektronensysteme und Gruppen gebildet und vernichtet werden, was, wie später zu sehen sein wird (siehe 6.6.1.6), auch auf die Atome zutrifft. So kann ein Teil der anfangs durchnumerierten Indizes im Laufe der Reaktionsschritte ungültig werden, weshalb die Möglichkeit besteht zu überprüfen, welche Indizes verfügbar sind. Die Aggregat- und Molekülindizes kann man nach jeder Teilreaktion über die Atome, die sie enthalten, abfragen.
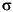 und
und 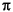 . Dabei gilt der Teil des Moleküls als eben, über das sich ein -System erstreckt. Sind
benachbarte -Systeme nicht miteinander konjugiert, zeigt dies eine Verdrehung des Moleküls aus der Ebene an dieser Stelle an. Neben den - und -Elektronensystemen besitzen die Moleküle
auch freie Elektronenpaare und unbesetzte Atomorbitale, die ebenfalls als -Systeme kodiert sind, nur an einem Atom beteiligt sind und 2 bzw. 0 Elektronen besitzen. Die Reaktionsgenerierung besteht nun darin, die richtigen -
und -Systeme zu finden und sie zu verbinden bzw. zu spalten. In den Regeln werden die Elektronensysteme angegeben, die ausgehend von den Atomen in der Reaktionssubstruktur gesucht werden. So kann beispielsweise das -System,
das die Atome 1 und 2 in der Reaktionssubstruktur verbindet, gebrochen werden. Hierzu stehen Eigenschaften, wie Elektronensysteme an einem bzw. zwischen zwei Atomen" zur Verfügung. Für den Bruch eines Elektronensystems wird angegeben, zwischen welchen
Atomen das Elektronensystem gespalten wird (siehe Atom A und B in Abbildung 88) und wieviele Elektronen dasjenige Elektronensystem erhält, an dem das Atom A beteiligt ist. -Systems-Systeme werden zwischen den beiden Atomen gespalten und es entstehen daraus zwei -Systeme, wobei auch freie Elektronenpaare als -Systeme kodiert sind (siehe oben). Das Elektronensystem,
an dem das Atom A beteiligt ist, erhält die angegebene Anzahl an Elektronen, das andere Elektronensystem die restlichen Elektronen des gespaltenen -Systems. Wird eine negative Elektronenzahl bzw. keine angegeben oder bleibt eine
negative Elektronenzahl für das andere Elektronensystem übrig, wird die Zahl der Elektronen automatisch so verteilt, daß jedes der entstehenden Elektronensysteme möglichst so viele Elektronen erhält, wie es zum nun gespaltenen Elektronensystem
beigetragen hat. Für diese automatische Verteilung der Elektronen werden intern Heuristiken (siehe 6.6.1.5) verwendet, deren Einsatz auf neutrale Moleküle oder Moleküle mit Oniumgruppen beschränkt ist, die
nur Hauptgruppenelemente ohne Oktettaufweitung und keine Mehrzentren--Systeme enthalten.
. Dabei gilt der Teil des Moleküls als eben, über das sich ein -System erstreckt. Sind
benachbarte -Systeme nicht miteinander konjugiert, zeigt dies eine Verdrehung des Moleküls aus der Ebene an dieser Stelle an. Neben den - und -Elektronensystemen besitzen die Moleküle
auch freie Elektronenpaare und unbesetzte Atomorbitale, die ebenfalls als -Systeme kodiert sind, nur an einem Atom beteiligt sind und 2 bzw. 0 Elektronen besitzen. Die Reaktionsgenerierung besteht nun darin, die richtigen -
und -Systeme zu finden und sie zu verbinden bzw. zu spalten. In den Regeln werden die Elektronensysteme angegeben, die ausgehend von den Atomen in der Reaktionssubstruktur gesucht werden. So kann beispielsweise das -System,
das die Atome 1 und 2 in der Reaktionssubstruktur verbindet, gebrochen werden. Hierzu stehen Eigenschaften, wie Elektronensysteme an einem bzw. zwischen zwei Atomen" zur Verfügung. Für den Bruch eines Elektronensystems wird angegeben, zwischen welchen
Atomen das Elektronensystem gespalten wird (siehe Atom A und B in Abbildung 88) und wieviele Elektronen dasjenige Elektronensystem erhält, an dem das Atom A beteiligt ist. -Systems-Systeme werden zwischen den beiden Atomen gespalten und es entstehen daraus zwei -Systeme, wobei auch freie Elektronenpaare als -Systeme kodiert sind (siehe oben). Das Elektronensystem,
an dem das Atom A beteiligt ist, erhält die angegebene Anzahl an Elektronen, das andere Elektronensystem die restlichen Elektronen des gespaltenen -Systems. Wird eine negative Elektronenzahl bzw. keine angegeben oder bleibt eine
negative Elektronenzahl für das andere Elektronensystem übrig, wird die Zahl der Elektronen automatisch so verteilt, daß jedes der entstehenden Elektronensysteme möglichst so viele Elektronen erhält, wie es zum nun gespaltenen Elektronensystem
beigetragen hat. Für diese automatische Verteilung der Elektronen werden intern Heuristiken (siehe 6.6.1.5) verwendet, deren Einsatz auf neutrale Moleküle oder Moleküle mit Oniumgruppen beschränkt ist, die
nur Hauptgruppenelemente ohne Oktettaufweitung und keine Mehrzentren--Systeme enthalten.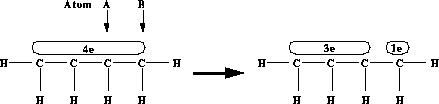 -Systems.-Systems wird das -System im ersten Schritt nicht gespalten, aber in der Datenstruktur als gebrochen markiert. Wird dieses -System an einer zweiten Stelle
oder alle - und -Systeme zwischen den Atomen gespalten, ist das ringförmige Elektronensystem dann aufgebrochen.-Systems-Systems entstehen zwei -Systeme, an denen je eines der beiden Atome beteiligt ist, also zwei nicht konjugierte p-Orbitale. Auch hier erhält das p-Orbital am Atom A die
angegebene Zahl von Elektronen und das andere den Rest. Würde für eines der p-Orbitale eine negative Elektronenzahl resultieren, zum Beispiel dadurch, daß die Elektronenverteilung nicht angegeben wurde, bekommt das p-Orbital am Atom A ein Elektron
und das am Atom B den Rest.
-Systems.-Systems wird das -System im ersten Schritt nicht gespalten, aber in der Datenstruktur als gebrochen markiert. Wird dieses -System an einer zweiten Stelle
oder alle - und -Systeme zwischen den Atomen gespalten, ist das ringförmige Elektronensystem dann aufgebrochen.-Systems-Systems entstehen zwei -Systeme, an denen je eines der beiden Atome beteiligt ist, also zwei nicht konjugierte p-Orbitale. Auch hier erhält das p-Orbital am Atom A die
angegebene Zahl von Elektronen und das andere den Rest. Würde für eines der p-Orbitale eine negative Elektronenzahl resultieren, zum Beispiel dadurch, daß die Elektronenverteilung nicht angegeben wurde, bekommt das p-Orbital am Atom A ein Elektron
und das am Atom B den Rest.  -Systems, wobei das Chloratom zwei Elektronen bekommt.-Systeme wird nur das Atom A aus dem -System herausgenommen und erhält ein p-Orbital mit der Elektronenzahl, die für diesen Teilschritt des Reaktionstyps in den Regeln festgelegt
wurde, bzw. null Elektronen.
-Systems, wobei das Chloratom zwei Elektronen bekommt.-Systeme wird nur das Atom A aus dem -System herausgenommen und erhält ein p-Orbital mit der Elektronenzahl, die für diesen Teilschritt des Reaktionstyps in den Regeln festgelegt
wurde, bzw. null Elektronen. -Systems.-Systems. Als Atom A wird das Metallatom angegeben, das durch den Bruch ein unbesetztes Atomorbital erhält, das aus der Komplexbindung entfernt
wird. So können nach und nach alle mit dem besetzten -System zu einer Komplexbindung vereinigten Atomorbitale herausgenommen werden, wodurch die Komplexbindung aufgehoben wird. Zurück bleiben schließlich die unbesetzten
Atomorbitale am Metall und das -System des Liganden., oder komplex sein. Ist kein Wert oder default" angegeben, wird ein -System erzeugt, wenn zwischen den Atomen kein -System existiert, anderenfalls ein
-System. Die Atome, zwischen denen die Elektronensysteme verknüpft werden, werden intern bestimmt, was für die Bildung von -Systemen keine zusätzliche Aufgabe ist. -Bindung und Konjugation von -Systemen-Bindung oder die Konjugation von -Systemen werden diese zu einem neuen, größeren vereinigt. Problemfälle, die hierbei auftreten können siehe 6.6.1.3.
-Systems.-Systems. Als Atom A wird das Metallatom angegeben, das durch den Bruch ein unbesetztes Atomorbital erhält, das aus der Komplexbindung entfernt
wird. So können nach und nach alle mit dem besetzten -System zu einer Komplexbindung vereinigten Atomorbitale herausgenommen werden, wodurch die Komplexbindung aufgehoben wird. Zurück bleiben schließlich die unbesetzten
Atomorbitale am Metall und das -System des Liganden., oder komplex sein. Ist kein Wert oder default" angegeben, wird ein -System erzeugt, wenn zwischen den Atomen kein -System existiert, anderenfalls ein
-System. Die Atome, zwischen denen die Elektronensysteme verknüpft werden, werden intern bestimmt, was für die Bildung von -Systemen keine zusätzliche Aufgabe ist. -Bindung und Konjugation von -Systemen-Bindung oder die Konjugation von -Systemen werden diese zu einem neuen, größeren vereinigt. Problemfälle, die hierbei auftreten können siehe 6.6.1.3.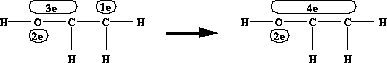 -Systeme zu einem neuen.-Systemen-Systeme werden aus zwei -Systemen gemacht. Am besten erzeugt man sie aus zwei -Systemen mit nur je einem Atom (p-Orbitalen). Zwischen diesen beiden Atomen wird dann das
-System gebildet und erhält die Summe der Elektronen der beiden p-Orbitale. Handelt es sich bei den -Systemen um Elektronensysteme mit mehr als einem Atom, werden zunächst die -Systeme
so gebrochen, daß an den Atomen ein Radikal (-System mit einem Atom und einem Elektron) entsteht. Da die Atome nicht übergeben werden, müssen sie intern bestimmt werden. Im Fall von zwei p-Orbitalen sind die Atome eindeutig,
aber bei ausgedehnteren -Systemen kann nicht immer gewährleistet werden, daß die richtigen Atome gefunden werden, obgleich versucht wird, die Atome mit minimaler Distanz in den beiden Elektronensystemen zu finden. Es empfiehlt
sich daher in den Regeln, -Systeme aus p-Orbitalen zu bilden und größere -Systeme vor der Bildung des -Systems selbst bis zur Bildung der benötigten p-Orbitale zu brechen.
-Systeme zu einem neuen.-Systemen-Systeme werden aus zwei -Systemen gemacht. Am besten erzeugt man sie aus zwei -Systemen mit nur je einem Atom (p-Orbitalen). Zwischen diesen beiden Atomen wird dann das
-System gebildet und erhält die Summe der Elektronen der beiden p-Orbitale. Handelt es sich bei den -Systemen um Elektronensysteme mit mehr als einem Atom, werden zunächst die -Systeme
so gebrochen, daß an den Atomen ein Radikal (-System mit einem Atom und einem Elektron) entsteht. Da die Atome nicht übergeben werden, müssen sie intern bestimmt werden. Im Fall von zwei p-Orbitalen sind die Atome eindeutig,
aber bei ausgedehnteren -Systemen kann nicht immer gewährleistet werden, daß die richtigen Atome gefunden werden, obgleich versucht wird, die Atome mit minimaler Distanz in den beiden Elektronensystemen zu finden. Es empfiehlt
sich daher in den Regeln, -Systeme aus p-Orbitalen zu bilden und größere -Systeme vor der Bildung des -Systems selbst bis zur Bildung der benötigten p-Orbitale zu brechen.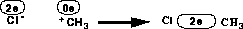 -Systems aus zwei -Systemen.-Systeme schrittweise gebrochen werden, werden sie auch schrittweise ausgehend von einem normalen 2-Elektronen-2-Zentren--System aufgebaut, wobei ein p-Orbital nach dem anderen hinzugefügt
wird. So entsteht nach und nach ein -System mit 3, 4, oder mehr Atomen.
-Systems aus zwei -Systemen.-Systeme schrittweise gebrochen werden, werden sie auch schrittweise ausgehend von einem normalen 2-Elektronen-2-Zentren--System aufgebaut, wobei ein p-Orbital nach dem anderen hinzugefügt
wird. So entsteht nach und nach ein -System mit 3, 4, oder mehr Atomen. 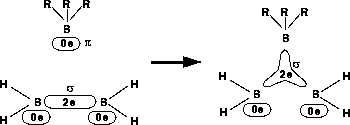 -Systems zum 3-Zentren--System.-Systeme als -Systeme ohne Zentrum, wie die BBB-Bindung in Abbildung 94. Soll ein -System, wie die BHB-Bindung,
gebildet werden, setzt man, nachdem man die 2-Elektronen-3-Zentren-Bindung erzeugt hat, mit der Eigenschaft EL_CENTER des Elektronensystems das Wasserstoffatom als Zentrum.
-Systems zum 3-Zentren--System.-Systeme als -Systeme ohne Zentrum, wie die BBB-Bindung in Abbildung 94. Soll ein -System, wie die BHB-Bindung,
gebildet werden, setzt man, nachdem man die 2-Elektronen-3-Zentren-Bindung erzeugt hat, mit der Eigenschaft EL_CENTER des Elektronensystems das Wasserstoffatom als Zentrum.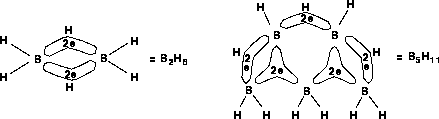 -System mit (BHB in B2H6 und B5H11) und ohne Zentrum (BBB in B5H11 [54]).-Bindungen werden auch Komplexbindungen ausgehend von einem -System und der entsprechenden Zahl leerer Atomorbitale am Metallatom schrittweise aufgebaut.-System herausgeschnitten werden. Die Schwierigkeit liegt
dabei in der Verteilung der Elektronen auf die entstehenden -Systeme. Dazu wird der gleiche Verteilungsmechanismus verwendet, der genommen wird, wenn eine negative Elektronenzahl für das Elektronensystem am ersten Atom bei einer
Spaltung angegeben wird (siehe 6.6.1.5). Diese arbeitet zuverlässig für neutrale Moleküle und Moleküle mit Oniumgruppen, in denen nur Hauptgruppenelemente enthalten sind, bei denen keine Oktettaufweitung
stattgefunden hat. Mehrzentren--Systeme und Komplexbindungen dürfen nicht Bestandteil der Moleküle sein.
-System mit (BHB in B2H6 und B5H11) und ohne Zentrum (BBB in B5H11 [54]).-Bindungen werden auch Komplexbindungen ausgehend von einem -System und der entsprechenden Zahl leerer Atomorbitale am Metallatom schrittweise aufgebaut.-System herausgeschnitten werden. Die Schwierigkeit liegt
dabei in der Verteilung der Elektronen auf die entstehenden -Systeme. Dazu wird der gleiche Verteilungsmechanismus verwendet, der genommen wird, wenn eine negative Elektronenzahl für das Elektronensystem am ersten Atom bei einer
Spaltung angegeben wird (siehe 6.6.1.5). Diese arbeitet zuverlässig für neutrale Moleküle und Moleküle mit Oniumgruppen, in denen nur Hauptgruppenelemente enthalten sind, bei denen keine Oktettaufweitung
stattgefunden hat. Mehrzentren--Systeme und Komplexbindungen dürfen nicht Bestandteil der Moleküle sein. Für alle Modifikationen muß intern auch noch mitprotokolliert werden, welche Elektronensysteme gelöscht wurden und welche neu gebildet wurden, damit von der Regel aus über die Indizes auch zugegriffen werden kann. Neue Elektronensysteme erhalten aufsteigend neue Indizes. In seltenen Fällen werden bei der Überarbeitung der chemischen Datenstruktur RICOS auch Gruppen erzeugt oder gelöscht, ohne daß die Verwaltung der Indizes etwas davon mitbekommt. Deshalb wird nach jedem neu gebildeten Produkt das gesamte Ensemble überprüft, ob sich Veränderungen daran ergeben haben.
-Systeme-Systeme erzeugt werden. In solchen Fällen werden die -Systeme vor
der Vereinigung automatisch so gespalten, daß dadurch unabhängige -Systeme entstehen und bei der Verknüpfung keine Probleme mehr vorhanden sind.
Abbildung 95 zeigt den letzten Teilschritt der Keto-Enol-Tautomerie von der Enol- in die Keto-Form. Zuvor wurden die C-C-Doppelbindung gespalten, wobei das eine Radikal mit dem freien Elektronenpaar vom Sauerstoffatom konjugiert
bleibt, die O-H-Bindung gespalten und die C-H-Bindung gebildet. Es fehlt die C-O-Bindung zur Bildung der Doppelbindung. Diese wird bevorzugt aus radikalischen, an einem Atom beteiligten Elektronensystemen erzeugt. Für das Sauerstoffatom wird dazu das Radikal
und am Kohlenstoffatom das Elektronensystem mit drei Elektronen gefunden. Da zwischen den beiden Atomen schon ein -System existiert, werden die beiden -Systeme miteinander verknüpft. Dies ist auch ganz
richtig, aber die Konjugation mit dem freien Elektronenpaar des Sauerstoffatoms muß dabei aufgehoben werden. Daß in jedem Fall beachtet wird, daß möglicherweise Konjugationen aufgehoben werden müssen, demjenigen aufzuerlegen, der eine
neue Regeln abfaßt, birgt die Gefahr, daß es vergessen wird. Deshalb wurde der Algorithmus zur Überprüfung und zum Ausschluß ungültiger -Systeme in EROS7 aufgenommen.
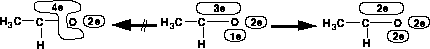 -Systems aus zwei anderen. Dies ist der letzte Teilschritt vom Enol- zu Keto-Form Tautomerie. -System entstehen würde. Es ist eine logische Fortentwicklung der Bildung eines ungültigen -Systems.
-Systems aus zwei anderen. Dies ist der letzte Teilschritt vom Enol- zu Keto-Form Tautomerie. -System entstehen würde. Es ist eine logische Fortentwicklung der Bildung eines ungültigen -Systems.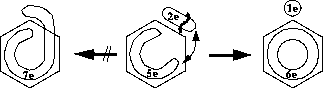 -Systems (links) bei der Kombination der beiden -Systeme wird dadurch ausgeschlossen, daß das kleinere -System zuerst gespalten
wird.
-Systems (links) bei der Kombination der beiden -Systeme wird dadurch ausgeschlossen, daß das kleinere -System zuerst gespalten
wird.
Soll eine Bindung zwischen zwei Atomen gebrochen werden, wird zunächst versucht, ein -System zu spalten. Ist kein -System zwischen den Atomen, wird das -System geteilt. Anschließend
werden die Elektronen auf die beiden entstandenen Elektronensysteme so verteilt, daß möglichst jedes Elektronensystem so viele Elektronen erhält, wie seine Atome zum nun gespaltenen Elektronensystem beigesteuert haben (siehe 6.6.1.5).
Elektronensysteme werden also homolytisch gespalten. Der Bruch eines Elektronensystems gilt dann als Bindungsbruch, wenn beide Elektronensysteme nach dem Bruch radikalisch sind. Ist dies nicht der Fall, wird angenommen, daß bei der Spaltung des Elektronensystems
nur eine Konjugation aufgehoben wurde. Soll eine Bindung gebrochen werden und wird festgestellt, daß nur eine Konjugation aufgehoben wurde, werden weiter Elektronensysteme gespalten, bis ein Bindungsbruch stattgefunden hat. Soll ein polarer Bindungsbruch
stattfinden und keine neue Bindungen gebildet werden, können nach dem homolytischen Bindungsbruch die Zahlen der freien Elektronen der beteiligten Atome um eins erhöht bzw. erniedrigt werden.
Existieren mehrere gleichwertige Grenzstrukturen in der Darstellung der Struktur als Bindungsliste, gibt es eine zusätzliche Betrachtungsweise. Nach dieser wird für die Spaltung eines -Systems zunächst immer diejenige
mesomere Grenzstruktur gewählt wird, bei der möglichst kein Bindungsbruch stattfindet (siehe Abbildung 97), um in einem weiteren Schritt dann die nächste Bindung (in Abbildung 97
die -Bindung) zu brechen.
 -Systems wird die Grenzstruktur gewählt, bei der nach Möglichkeit kein Bindungsbruch eintritt. Die gestrichelte Linie deutet den Ort der Spaltung des -Systems
an.-System aufgebrochen, gilt dies normalerweise als Bindungsbruch, da sonst mit einem einzigen Bruch in einem Aromaten sowohl das -System als auch die -Bindung im Ring
gespalten würden. Möchte man dies nicht, kann man dies beim Bindungsbruch angeben.
-Systems wird die Grenzstruktur gewählt, bei der nach Möglichkeit kein Bindungsbruch eintritt. Die gestrichelte Linie deutet den Ort der Spaltung des -Systems
an.-System aufgebrochen, gilt dies normalerweise als Bindungsbruch, da sonst mit einem einzigen Bruch in einem Aromaten sowohl das -System als auch die -Bindung im Ring
gespalten würden. Möchte man dies nicht, kann man dies beim Bindungsbruch angeben.
Für die Bildung einer Bindung werden an den Atomen bevorzugt radikalische Elektronensysteme herangezogen. Das Radikalzentrum kann dabei konjugiert oder nicht konjugiert sein. Ein -System wird zwischen zwei Atomen generiert, wenn
zwischen diesen Atomen noch keines existiert, wobei zunächst aus den radikalischen -Systemen falls nötig die p-Orbitale herausgenommen werden und daraus dann das -System gebildet wird. Ist zwischen
den Atomen schon ein -System vorhanden, werden die beiden radikalischen -Systeme vereinigt, wobei auch die möglichen Fälle der Bildung ungültiger -Systeme (siehe 6.6.1.3)
berücksichtigt werden.
Im Laufe der Reaktionen werden so nach und nach auch die Konjugationen aufgehoben. Um Moleküle nicht doppelt oder dreifach zu erzeugen (mit und ohne Konjugationen), müssen die -Systeme nach jeder Reaktion wieder konjugiert
werden. Der Hashcoder, der für die Erkennung identischer Moleküle und Molekülteile zuständig ist, unterscheidet Moleküle, deren -Systeme konjugiert sind bzw. nicht. Er wurde für die MO-orientierte Datenstruktur
RICOS geschrieben, bei der dieser Unterschied eine Verdrehung im Molekül anzeigt.
Die automatische Verteilung der Elektronen auf die beiden Elektronensysteme bei der Spaltung eines -Systems geht von einer Tabelle aus, in der die normale Zahl von unkonjugierten Orbitalen - unbesetzt, teilweise besetzt oder besetzt
- vermerkt ist. Diese Zahl ist natürlich von der Zahl der Orbitale abhängig, die ein Atom der verschiedenen Elemente in der Darstellung nach RICOS besitzt und ist für die verschiedenen Elemente in der folgenden Tabelle angegeben:
H He
0, 1,
Li Be B C N O F Ne
0, 0, -1, 0, 1, 2, 3, 4,
Na Mg Al Si P S Cl Ar
0, 0, -1, 5, 6, 7, 8, 9,
K Ca Sc Ti V Cr Fe Mn Co Ni Cu Zn Ga Ge As Se Br Kr
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,-1, 5, 6, 7, 8, 9,
Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,-1, 5, 6, 7, 8, 9,
Cs Ba La
0, 0, 0,
Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn
0, 0, 0, 0, 0, 0, 0, 0, 0,-1, 5, 6, 7, 8, 9,
Fr Ra Ac
0, 0, 0,
Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
Rf Ha ......
0, 0, 0, 0, 0, 0, 0, 0, 0,-1, 5, 6, 7, 8, 9
Die Elemente, die Elektronenmangelverbindungen bilden, haben den Wert minus eins, der besagt, daß Atome dieser Elemente keine Elektronen zu einem -System beitragen, wenn eine Verbindung tatsächlich einmal an einem -System
beteiligt sein sollte. Für die Hauptgruppen IV, V, VI und VII sind die Zahlen so gesetzt, daß sie 4, 3, 2 und 1 Bindung eingehen und die restlichen Orbitale in der RICOS-Darstellung als freie Elektronenpaare oder leere Orbitale besitzen. So ist für
Stickstoff der Normalzustand mit einem freien Elektronenpaar kodiert, wodurch er drei Bindungen eingeht. Phosphor hat die 6, da er bei RICOS als Atom neun und nicht wie der Stickstoff vier Orbitale besitzt. Daraus ergibt sich als Normalzustand für den
Phosphor eine Anzahl von einem freien Elektronenpaar und fünf leeren Orbitalen neben drei Bindungen. In dieser Tabelle sind die höheren Homologen also ohne Oktettaufweitung kodiert.
Zur Bestimmung, wieviele Elektronen normalerweise in den beiden Teilen eines gespaltenen -Systems enthalten sein sollten, wird für beide Elektronensysteme für jedes Atom die Zahl der Elektronen aufsummiert, die es beisteuert.
Wieviele Elektronen kommen nun von einem Atom? Dazu werden zunächst die verschiedenen Elektronensysteme an diesem Atom gezählt. Man erhält die Zahlen der konjugierten -Systeme, zu denen das gespaltene -System
immer dazuzählt (auch, wenn es ein p-Orbital wurde), die Zahl der nicht konjugierten Orbitale (z.B. freie Elektronenpaare) und die Zahl der -Systeme. Bei dieser Zählung ist die Besetzung der einzelnen Elektronensysteme unerheblich.
Das Atom steuert dann zwei Elektronen zum -System bei, wenn das Atom weniger unkonjugierte Orbitale hat als in der Tabelle eingetragen sind, ansonsten beteiligt es sich mit einem Elektron.
Für einige Elemente lassen sich aber auch globale Aussagen machen: Elemente der dritten Hauptgruppe donieren kein Elektron, die Edelgase donieren zwei Elektronen und Wasserstoff, die Elemente der vierten Hauptgruppe, die Alkali- und Erdalkalimetalle sowie
alle Nebengruppenelemente, Lanthaniden und Actiniden ein Elektron. Zu beachten ist überdies, daß die Oniumatome jeweils zwei Elektronen zum -System beitragen, die sie erhalten würden, wenn die Konjugation aufgehoben würde.
In Fällen, in denen kein konjugiertes Elektronensystem am betrachteten Atom ist, wird dieser Algorithmus nicht verwendet.
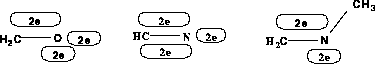 -System doniert.
-System doniert. -System doniert.
-System doniert. -System doniert (Oniumatome; die Formalladung ist nicht Bestandteil der chemischen Datenstruktur RICOS, sondern wurde nur zur besseren Lesbarkeit angegeben.).-System mehr oder weniger Elektronen als die so bestimmte Summe der normalen Elektronenzahlen der beiden neuen Elektronensysteme, erhält die Differenz - Defizit oder Überschuß - das Elektronensystem,
das mehr Elektronen zugeteilt bekommen hat.
-System doniert (Oniumatome; die Formalladung ist nicht Bestandteil der chemischen Datenstruktur RICOS, sondern wurde nur zur besseren Lesbarkeit angegeben.).-System mehr oder weniger Elektronen als die so bestimmte Summe der normalen Elektronenzahlen der beiden neuen Elektronensysteme, erhält die Differenz - Defizit oder Überschuß - das Elektronensystem,
das mehr Elektronen zugeteilt bekommen hat.
Mit dieser Methode erhält man für neutrale Verbindungen aus Hauptgruppenelementen ohne Oktettaufweitung und Oniumverbindungen die richtige Zahl von Elektronen, wenn sie keine Mehrzentren--Systeme und Komplexbindungen enthalten.
Einen Spezialfall stellt noch ein Stickstoffatom mit zwei -Systemen und zwei konjugierten -Systemen dar. In diesem Fall doniert das Stickstoffatom in das eine -System zwei und in das
andere ein Elektron. Da auch mehrere solcher Atome im Molekül enthalten sein können und sich die -Systeme auch noch überschneiden können, kann nicht zuverlässig festgelegt werden, in welches -System
wieviele Elektronen doniert werden, zumal es sich in der Praxis auch um Radikale, Kationen und Radikalkationen handeln kann. Deshalb wurde am obigen Schema keine Korrektur für diesen Fall vorgenommen. Damit erhält das betrachtete -System
für dieses Atom zwei Elektronen. Wird dieses -System dann gespalten, erhält das noch verbleibende -System ein Elektron.
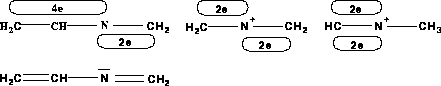 -System zwei Elektronen
(freies Elektronepaar) und eines in das recht -System der Doppelbindung zur Methylengruppe.
-System zwei Elektronen
(freies Elektronepaar) und eines in das recht -System der Doppelbindung zur Methylengruppe.Die Gruppen dienen sowohl zum Markieren, wie im Fall der funktionellen Gruppen, als auch zur Bildung von Aggregaten. Wird eine solche Gruppe erzeugt, beschreibt sie eine spezielle Wechselwirkung, wie die Wasserstoffbrückenbindung. Faßt sie Atome aus verschiedenen Molekülen zusammen, bilden die Moleküle ein Aggregat, also ein Teilchen für EROS7. Die Quartärstruktur eines Proteins wäre ein Aggregat, das aus den Proteinsträngen, den Molekülen, besteht. Ob nun eine Gruppe in der Lage ist, ein Aggregat zusammenzuhalten oder gelöscht bzw. geteilt wird, wenn die Atome und/oder Elektronensysteme in verschiedenen Molekülen zu liegen kommen, wird angegeben, wenn diese Gruppe erzeugt wird.
Eine Möglichkeit eine Wasserstoffbrückenbindung zu kodieren wäre, die zwei Atome, das Wasserstoffatom und den Elektronendonor sowie das freie Elektronenpaar des Donoratoms in einer Gruppe zusammenzufassen und sie h-bridge zu nennen und diese dem Aggregat zuzuordnen.
 -Systems oder etwaiger Stereodeskriptoren, alle genealogischen Variablen. Wird eine genealogische Variable beim
Initialisieren der Regeln mit einem Namen sowie dem Daten- und Chemietyp angemeldet, kann sie für alle Ensembles bei der Überprüfung der Bedingungen gelesen und im Funktionsteil selbst gelesen und gesetzt werden. Als Datentypen stehen ganzzahlige
Werte (int), Fließkommazahlen (double), Vektoren aus diesen beiden sowie Zeichenketten und 64-Bit-Werte für Hashcodes zur Verfügung. RICOS kennt noch weitere Datentypen, die aber von der Regelschnittstelle auf diese sechs Datentypen abgebildet
werden. Der Chemietyp gibt an, zu welchen Objekten diese genealogische Variable gehört. Das sind Atome, Elektronensysteme, Gruppen, Moleküle und Aggregate. Wird sie gelesen, ohne daß ihr zuvor ein Wert zugewiesen wurde, erhält man einen
default-Wert, der in der Regel null ist. Die genealogischen Variablen dienen in ihrer Hauptsache dazu, die Geschichte der Entstehung eines Aggregates zu speichern. In der Massenspektroskopie werden die Moleküle bei der Ionisation gleichzeitig auch angeregt.
Im Laufe der Fragmentierungen verliert das Kation meist stark an Anregungsenergie. Mit einer genealogischen Variable, in der man bei jeder Reaktion die Reaktionsenthalpie auf die Anregungsenergie nach der Ionisation addiert und die resultierende Anregungsenergie
auf das Kation und das Neutralfragment aufteilt, erhält man für jedes Kation seine Anregungsenergie. Da oft große Neutralfragmente mit vielen Freiheitsgraden abgespalten werden, verliert das Kation in nur wenigen Reaktionsschritten fast seine
komplette Anregungsenergie. Reaktionen mit einer hohen Aktivierungsbarriere können so für kalte Ionen ausgeschlossen werden.
-Systems oder etwaiger Stereodeskriptoren, alle genealogischen Variablen. Wird eine genealogische Variable beim
Initialisieren der Regeln mit einem Namen sowie dem Daten- und Chemietyp angemeldet, kann sie für alle Ensembles bei der Überprüfung der Bedingungen gelesen und im Funktionsteil selbst gelesen und gesetzt werden. Als Datentypen stehen ganzzahlige
Werte (int), Fließkommazahlen (double), Vektoren aus diesen beiden sowie Zeichenketten und 64-Bit-Werte für Hashcodes zur Verfügung. RICOS kennt noch weitere Datentypen, die aber von der Regelschnittstelle auf diese sechs Datentypen abgebildet
werden. Der Chemietyp gibt an, zu welchen Objekten diese genealogische Variable gehört. Das sind Atome, Elektronensysteme, Gruppen, Moleküle und Aggregate. Wird sie gelesen, ohne daß ihr zuvor ein Wert zugewiesen wurde, erhält man einen
default-Wert, der in der Regel null ist. Die genealogischen Variablen dienen in ihrer Hauptsache dazu, die Geschichte der Entstehung eines Aggregates zu speichern. In der Massenspektroskopie werden die Moleküle bei der Ionisation gleichzeitig auch angeregt.
Im Laufe der Fragmentierungen verliert das Kation meist stark an Anregungsenergie. Mit einer genealogischen Variable, in der man bei jeder Reaktion die Reaktionsenthalpie auf die Anregungsenergie nach der Ionisation addiert und die resultierende Anregungsenergie
auf das Kation und das Neutralfragment aufteilt, erhält man für jedes Kation seine Anregungsenergie. Da oft große Neutralfragmente mit vielen Freiheitsgraden abgespalten werden, verliert das Kation in nur wenigen Reaktionsschritten fast seine
komplette Anregungsenergie. Reaktionen mit einer hohen Aktivierungsbarriere können so für kalte Ionen ausgeschlossen werden.
Der gleichen Gedankenansatz kann dazu führen, daß kein Interesse an einem der Produkte besteht, nachdem eine Reaktion vollständig durchgeführt wurde. Denkbar wären hier das abgespaltene Neutralfragment bei massenspektroskopischen Prozessen, die nicht detektiert werden können, wodurch auch ihre entstandene Menge uninteressant ist. Deshalb ist es möglich, während der Reaktion auch ganze Aggregate aus dem Ensemble zu löschen. Weil über die Eigenschaften zunächst abgefragt werden muß, welche Nummer das zu löschende Aggregat hat, kann das Löschen des Aggregates erst als zweiter Teilschritt durchgeführt werden. Auch hier sind die ausgegebenen Reaktionen nicht stöchiometrisch korrekt. Im Falle der Massenspektroskopie gibt es jedoch eine bessere Möglichkeit, die Neutralfragmente von einer Weiterreaktion auszuschließen und dennoch stöchiometrisch korrekte Reaktionen zu erhalten (siehe 6.4.2).
Die Einführung von Edukten während Reaktionen pseudoerster Ordnung wurde auf diese Weise durchgeführt, damit die Ordnung der Reaktion und, von welcher Stoffkonzentration die Reaktionsgeschwindigkeit abhängt, zur Aufstellung der Differentialgleichungen automatischen abgeleitet werden können. Wird darauf bestanden, stöchiometrisch korrekte Reaktionen zu formulieren, besteht die Möglichkeit, die Reaktion als Reaktion zweiter Ordnung zu formulieren, wobei die Konzentrationen der Edukte entsprechend gesetzt werden. Die Simulation mit Reaktionen zweiter Ordnung ist langsamer, da EROS7 mehrere Eduktkombinationen durchtesten muß, bevor es die richtige gefunden hat.
Als Beispiel sind in Abbildung 104 die n- und -Ionisationen von Acrylsäure angebenen. Würde der Reaktionstyp der Ionisation ein Atom im Reaktionszentrum enthalten, würde die Ionisation
des konjugierten -Systems mit sechs Elektronen für jedes der fünf an ihm beteiligten Atome erzeugt. Die Reaktionssubstruktur ohne Atome wird allerdings nur ein einziges Mal in jedem beliebigen Molekül gefunden und damit
auch nur einmal für Acrylsäure. Um alle drei in Abbildung 104 gezeigten Ionisationen durchführen zu können, ist es möglich aus der einen gefunden Reaktionssubstruktur ohne Atome mehrere Reaktionen zu
generieren, nämlich alle drei Ionisationen.
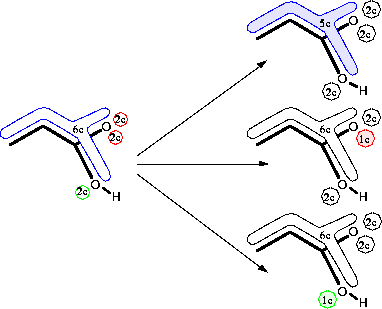 ) von Acrylsäure. Die Elektronensysteme, aus denen ein Elektron enfernt wurde, sind grau markiert.
) von Acrylsäure. Die Elektronensysteme, aus denen ein Elektron enfernt wurde, sind grau markiert.Die Eigenschaften werden abgefragt, wenn das entsprechende Ensemble aktiv ist. Zu Beginn der Reaktion können die Eigenschaften des Eduktensembles abgefragt und bereits der Eduktfunktionsteil berechnet werden. Ist ein Reaktionsteilschritt bzw. die Gesamtreaktion abgeschlossen, können die Eigenschaften von diesem Ensemble (einem Übergangszustand, Zwischenprodukt oder dem Produkt) der Reaktion, abgeholt und zur Reaktivität verrechnet werden.
Werden während der Reaktion jedoch Moleküle eingeschleust, muß die Symmetriezahl der Reaktion mit der der reagierenden Substruktur im eingeschleusten Molekül multipliziert werden. Im Fall der Ionisationsreaktion (siehe Erzeugung mehrerer Reaktionen aus einer gefundenen RSS auf Seite 97) wird die Symmetriezahl auf den Wert gesetzt, der angibt wieviele gleiche Elektronensysteme im Molekül vorhanden sind. Diese Zahl kann als Eigenschaft des Elektronensystems abgefragt werden.
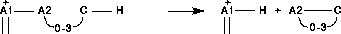
Generell ist es hier möglich, die Reaktionen, die von einem Eduktensemble ausgehen, zu vergleichen und die Reaktivitätswerte der Reaktionen gegebenenfalls anzupassen oder eine Reaktion ganz zu verwerfen. Dazu stehen alle an das Kernsystem von EROS7 übergebenen Variablen der Typen int und double sowie die Symmetriezahl der Reaktionen, die Phase für die Produkte (siehe unten) und eine Nummer für die Entscheidungen zur Verfügung, aus der die Identität der Produkte erkannt werden kann. Die Nummer 1 steht dabei für das Eduktensemble und alle anderen Nummern stehen für neue Produkte, wobei gleiche Produkte auch gleiche Nummern erhalten. Auf die Aggregate selbst kann jedoch nicht zugegriffen werden. Bei einem komplexen Satz von Reaktionstypen, wie sie für die Analyse und Simulation von Massenspektren (siehe 4.5.3) verwendet wurden, kann es vorkommen, daß eine Reaktion vom Edukt zum Produkt von zwei verschiedenen Reaktionstypen erzeugt wird (siehe Abbildung 106).

 -Spaltung und der Halogenspaltung. Die gestrichelte Linie zeigt die Spaltung der Bindung an.
-Spaltung und der Halogenspaltung. Die gestrichelte Linie zeigt die Spaltung der Bindung an.Für organische Reaktionen ist die Verteilungsfunktion ohne Bedeutung. Hier genügt die in EROS7 eingebaute Verteilungsfunktion, die aufgerufen wird, wenn in den Regeln keine angegeben ist. Sie verwirft Reaktionsduplikate und Reaktionen, die das Edukt nicht verändern und keine Phasenübergänge sind.
Schließlich können hier zusätzliche Werte für die Reaktionen berechnet werden.