![[Prev]](p_prev.gif)
![[Next]](p_next.gif)
RULE_INFO
CONSTR
FUNC
In RULE_INFO werden der Regelname, die Regelattribute und die Reaktionssubstruktur angegeben. Die Überprüfung der Bedingungen an der Reaktionssubstruktur findet im Teil CONSTR statt. Durchgeführt wird die Reaktion schließlich
in FUNC inklusive der Berechnung der Reaktivitätsfunktion aus Edukt- und Produktparametern. Der Aufbau einer Regel sieht dann wie folgt aus:
// Regel Nummer 1
case 1:
switch (op) {
case RULE_INFO:
{
// Reaktionssubstruktur, Name, Attribute
return OK;
}
case CONSTR:
// Bedingungen an der Reaktionssubstruktur
return OK;
case FUNC:
// Reaktionsfunktion
return OK;
default:
return BAD;
}
return OK;
RULE_1 {
switch $op {
RULE_INFO {
# Reaktionssubstruktur, Name, Attribute
}
CONSTR {
# Bedingungen an der Reaktionssubstruktur
}
FUNC {
# Reaktionsfunktion
}
default {
return BAD
}
}
return OK
}
NULL-terminiertes const char-Array mit den Attributen der Regel. Die Attribute der Regel werden dazu verwendet, einen Reaktionstyp für die Wahrscheinlichkeitskinetik als Umlagerungsreaktionstyp
(rearrangement) zu kennzeichnen oder für sie eine Michaelis-Menten-Kinetik (michaelis_menten_kinetics) bzw. eine Kinetik nullter Ordnung (force_zero_order) festzulegen. Ist ein entsprechendes trace-Level gewählt,
werden die Attribute und der Name für die einzelnen Regeln ausgegeben.
val->putco("rule_name", "Enzymreaktion");
static const char *const attrib_al[]={"michaelis_menten_kintetics",NULL};
val->putco("attributes", attrib_al);
global attrib_1 rule_1 set rule_1 "Enzymreaktion" putco rule_name rule_1 set attrib_1(0) michaelis_menten_kintetics set attrib_1(1) NULL putco attributes attrib_1
RULE_INFO ist die Kodierung der Reaktionssubstruktur (RSS). Die Reaktionssubstruktur kann aus 0 bis beliebig vielen Teilzentren bestehen, die wiederum beliebig verzweigt sein können. Auch Ringe sind in der Reaktionssubstruktur
zugelassen. Da die Reaktionssubstruktur so freizügig gestaltet ist, wird sie als Satz von Nachbarschaftsbeziehungen beschrieben. Die Atome in der Reaktionssubstruktur sind von 1 bis N durchnumeriert. Sie werden im übrigen auch in dieser Reihenfolge
gesucht, was für die Numerierung in der RSS von Bedeutung ist. Aus Effizienzgründen empfiehlt es sich, den spezifischsten Atomen in der RSS die niedrigsten Nummern zu geben.Die Kodierung der RSS ist ein 0-terminiertes Array aus Zahlentrippeln:
a1, a2, d
a1 und a2 sind die Nummern der Atome in der RSS, wobei die Reihenfolge ohne Bedeutung ist. d ist für Erweiterungen reserviert und kann im Moment nur die Werte 0 oder 1 annehmen, die beide
bedeuten, daß die Atome a1 und a2 direkt benachbart sind. Ursprünglich war gedacht, daß man mit d auch einen größeren Abstand zwischen den Atomen angeben kann. Es ist aber flexibler, dies über
Eigenschaften zu lösen und die Überprüfung im Teil CONSTR vorzunehmen. Für eine 3-atomige RSS 1-2-3 sieht der Code folgendermaßen aus:
static const int center_al[]={ 1, 2, 0, 2, 3, 0, 0 };
val->putco("center_connectivity", center_al);
global center_1
set center_1 {1,2,0,2,3,0,0}
putco center_connectivity center_1
Die Atome eines Teilzentrums sollten in einer Reihe durchnumeriert sein, wobei die Teilzentren automatisch erkannt werden, da keine Atome aus verschiedenen Teilzentren Nachbarschaftsbeziehungen besitzen. Will man explizit ein einzelnes Atom als Teilzentrum
spezifizieren, kann man das mita3, a3, 0tun. Dies ist nur nötig, wenn es das letzte Atom in der RSS ist.
CONSTR einer Regel werden die Bedingungen, die an eine RSS über die reine Konnektivität hinaus gestellt werden, überprüft. Dies geschieht ebenso schrittweise, wie das Reaktionszentrum gesucht wird. Als Indikator, wieviele
Atome im aktuellen Reaktionszentrum schon gefunden wurden, dient dabei der Übergabeparameter sub_op, der in Tcl implizit definiert ist. Für eine Reaktion wird die Funktion CONSTR nacheinander mit sub_op = 0, 1, 2,
... aufgerufen, bis alle Atome in der RSS überprüft wurden. Ist sub_op = 0, können die Eigenschaften des Ensembles, der Aggregate und Moleküle getestet werden. Für die Werte 1, 2, ... sind entsprechend viele Atome der RSS
gefunden. Sind zwei Atome gefunden, können alle Eigenschaften der Atome 1 und 2 überprüft werden. Es empfiehlt sich allerdings, die Überprüfungen, die nicht vom Atom 2 abhängen, bereits für sub_op = 1 durchzuführen.
Benötigt man Oderverknüpfungen zwischen Eigenschaften der Atome 1 und 2, können diese erst überprüft werden, wenn auch das zweite Atom gefunden wurde.
switch (sub_op) {
case 0: {
// Überprüfung des Eduktensembles
break;}
case 1:
// Überprüfung von Atom 1 in der RSS
break;}
case 2:
// Überprüfung von Atom 2
// sowie Oderbedingungen von Atom 1 und 2
break;}
case 3:
// Überprüfung von Atom 3
// sowie Oderbedingungen der Atome 3 und 1 sowie 3 und 2
break;}
default:
return BAD;
}
return OK;
switch $sub_op {
0 {
# Überprüfung des Eduktensembles
}
1 {
# Überprüfung von Atom 1 in der RSS
}
2 {
# Überprüfung von Atom 2
# sowie Oderbedingungen von Atom 1 und 2
}
3 {
# Überprüfung von Atom 3
# sowie Oderbedingungen der Atome 3 und 1 sowie 3 und 2
}
default {
return BAD
}
}
Die einzelnen Teile von CONSTR für die unterschiedliche Anzahl gefundener Atome der verschiedenen Regeln werden im übrigen für die Regeln und die Reaktionen durcheinander aufgerufen. Es ist also fast ausgeschlossen, sich Werte zu merken, wenn
ein Atom gefunden wurde, und diese zu verwenden, wenn auch ein weiteres Atom gefunden wurde.CONSTR die Eigenschaften der Edukte überprüft werden. Dazu müssen zunächst die entsprechenden Eigenschaften abgefragt
werden. Zu den Eigenschaften gehören auch Angaben über die Beschaffenheit der chemischen Struktur. Die Abfrage geschieht mit den Funktionen:bool flag; var_type variable; flag = eifc->prop(variable, prop_name, index_1, index_2);
set flag [prop prop_name index_1 index_2 variable]Abhängig von der Eigenschaft
prop_name entfallen die Indizes teilweise. Die überwiegende Zahl aller Eigenschaften außer derer des ganzen Ensembles und derer von Nachbaratomen haben nur einen Index (Aggregat, Molekül, Gruppe,
Elektronensystem und Atom). index_2 entfällt in diesem Fall. Ensembleeigenschaften haben gar keinen Index und Nachbarschaftseigenschaften beide Indizes. Der Rückgabewert der Funktionen prop ist ein Flag, das angibt, ob die
Eigenschaft erfolgreich abgeholt werden konnte (true = erfolgreich, Tcl: 1).Die Indizes sind dabei diejenigen des Ensembles. Die Angabe des Index 1 für eine Atomeigenschaft bedeutet, daß diese Eigenschaft für das Atom 1 im Ensemble angefordert wird. Will man die Eigenschaft des ersten Atoms in der RSS, so muß man sich zunächst den Index dieses Atoms holen. Diese etwas umständlichere Methode ist durchaus beabsichtigt, da man so das ganze Ensemble im Zugriff hat und, wenn gewünscht, auch die Atome usw. in der Nachbarschaft der RSS überprüfen kann.
Das folgende Beispiel zeigt, wie man sich die Ordnungszahl des ersten Atoms in der RSS abholen kann.
bool flag;
int atom_index, elem;
atom_index = eifc->center(1);
flag = eifc->prop(elem, "A_ELEMENT", atom_index);
if (flag) {
print << "Das Atom 1 in der RSS ist " << atom_index;
print << " und hat die Ordnungszahl " << elem << endl;
}
set atom_index [center 1]
set flag [prop A_ELEMENT $atom_index elem]
if {$flag} {
print "Das Atom 1 in der RSS ist $atom_index"
print " und hat die Ordnungszahl $elem\n"
}
In C++ kann der Name der Eigenschaft wahlweise als const char* ( "A_ELEMENT" ) angegeben werden oder man macht sich allgemein ein Objekt der Klasse rx_prent, das dann immer wieder verwendet werden kann. Dieses Objekt wird
dann statt des Eigenschaftsnamens übergeben und mit dem Konstruktor erzeugt, der den Namen als const char* erwartet. Da intern nicht bei jeder Abfrage die Eigenschaft über einen Stringvergleich in mehreren Listen gesucht werden muß,
ist die etwas schnellere Methode in C++ eine Eigenschaft abzufragen diejenige über die Erzeugung eines Objekts der Klasse rx_prent.
static rx_prent ordnz("A_ELEMENT");
bool flag;
int atom_index, elem;
atom_index = eifc->center(1);
flag = eifc->prop(elem, ordnz, atom_index);
if (flag) {
print << "Das Atom 1 in der RSS ist " << atom_index;
print << " und hat die Ordnungszahl " << elem << endl;
}
In C++ muß die Variable vom richtigen Typ sein, wobei es Eigenschaften mit folgenden Typen gibt:
bool prop( int &var, const char* prop_name, int idx1=0, int idx2=0); bool prop( double &var, const char* prop_name, int idx1=0, int idx2=0); bool prop( vector<int> &var, const char* prop_name, int idx1=0, int idx2=0); bool prop(vector<double> &var, const char* prop_name, int idx1=0, int idx2=0); bool prop( e_bit64 &var, const char* prop_name, int idx1=0, int idx2=0); bool prop( const char * &var, const char* prop_name, int idx1=0, int idx2=0); bool prop( int &var, const rx_prent &p, int idx1=0, int idx2=0); bool prop( double &var, const rx_prent &p, int idx1=0, int idx2=0); bool prop( vector<int> &var, const rx_prent &p, int idx1=0, int idx2=0); bool prop(vector<double> &var, const rx_prent &p, int idx1=0, int idx2=0); bool prop( e_bit64 &var, const rx_prent &p, int idx1=0, int idx2=0); bool prop( const char * &var, const rx_prent &p, int idx1=0, int idx2=0); rx_prent(const char *name);Darüber hinaus gibt es noch ein paar weitere nützliche Funktionen, mit deren Hilfe man feststellen kann, welche Eigenschaften verfügbar sind und welche Typen sie besitzen:
const char* prop_name(int idx); const char* prop_type(const char *prop_name); int prop_idxs(const char *prop_name); rx_prent(int idx);In Tcl gibt es keine Unterscheidung bezüglich der Variablentypen und damit auch nur eine Funktion.
prop property_name $idx1 $idx2 variable_name set property_name prop_name($idx) set ptype prop_type($property_name) set idxs prop_idxs($property_name)Der Name der Variablen kann in Tcl entfallen. Er ist dann identisch mit dem Namen der Eigenschaft.
Die so erhaltenen Eigenschaften können überprüft und verrechnet werden, wodurch entschieden wird, ob die (Teil-)Substruktur für die angestrebte Reaktion geeignet ist oder nicht.
Im folgenden Beispiel muß es sich bei der Überprüfung von Atom 1 in der RSS um ein Kohlenstoffatom handeln.
bool flag; int atom_index, element; atom_index = eifc->center(1); flag = eifc->prop(elem, "A_ELEMENT", atom_index); if (elem != 6) return BAD; return OK;
set atom_index [center 1]
set flag [prop A_ELEMENT $atom_index elem]
if {$elem != 6} {return BAD}
return OK
change_bond_order und change_free_els. Bei der Änderung der Bindungsordnung sind ia1
und ia2 (siehe unten) die Indizes der Atome, zwischen denen die Bindungsordnung geändert werden soll und delta die Änderung. Ist delta größer als null, werden entsprechend viele Bindungen erzeugt, für
delta < 0 gebrochen. Das optionale Flag single_aro_split gibt schließlich noch an, ob der einfache Bruch eines cyclischen 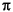 -Systems als Bindungsbruch gewertet werden soll (
-Systems als Bindungsbruch gewertet werden soll (=1) oder nicht
(=0). Ist single_aro_split = 0 und spaltet man in Benzol eine C-C-Bindung, wird die bevorzugt durchgeführt Spaltung des -Systems nicht als Bindungsbruch interpretiert. Dies hat zur Folge, daß ein weiteres
Elektronensystem gespalten wird, jetzt das 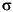 -System, und der Benzolring ist somit an dieser Stelle komplett aufgebrochen worden. In der Regel will man aber das -System des Aromaten an zwei Stellen aufbrechen
und die C-C--Bindungen intakt lassen. Deshalb ist der default-Wert für
-System, und der Benzolring ist somit an dieser Stelle komplett aufgebrochen worden. In der Regel will man aber das -System des Aromaten an zwei Stellen aufbrechen
und die C-C--Bindungen intakt lassen. Deshalb ist der default-Wert für single_aro_split = 1. So wird im ersten Schritt das cyclische -System zwar nicht gebrochen, aber zum Bruch markiert.
Im zweiten Schritt bricht das cyclische -System dann unabhängig von single_aro_split in zwei Teile. change_bond_order erkennt auch selbstständig Konjugationen und hebt diese gegebenenfalls auf, bevor
der eigentliche Bindungsbruch erfolgt. Dabei wird folgendermaßen vorgegangen: es wird zunächst ein -System (falls vorhanden) gespaltet und überprüft, ob es sich dabei um einen homolytischen Bindungsbruch gehandelt
hat. Dies ist der Fall, wenn beide resultierenden Elektronensysteme eine ungerade Zahl an Elektronen haben, wobei die Elektronen so verteilt werden, daß nach Möglichkeit so viele Elektronen in beide Elektronensysteme gegeben werden, wie die nun am
Elektronensystem beteiligten Atome zum ursprünglichen Elektronensystem beigetragen haben. Da auf diese Weise auch Konjugationen aufgehoben werden können, aber entstehende Lonepairs und -Systeme nicht automatisch konjugiert
werden, gibt es die Funktion conjugate_elsys. Sie konjugiert die entstandenen Elektronensysteme nach Möglichkeit, was während der einzelnen Reaktionsschritte hinderlich wäre, da für die nächsten Reaktionsschritte die Konjugation
höchstwahrscheinlich wieder aufgehoben werden müßte. Außerdem gibt es uneindeutige Fälle, in denen aufgrund von Heuristiken eine der Möglichkeiten gewählt wird, die aber im Einzelfall nicht der gewünschten entsprechen
muß. So besteht die Möglichkeit, die Elektronensysteme selbst zu konjugieren oder dies von der Funktionen conjugate_elsys automatisch erledigen zu lassen. Funktionen, die ein Flag zurückgeben, kommen beim Fehlschlagen mit false
bzw. 0 in Tcl zurück. Wird die Funktion conjugate_elsys nach Abschluß der Reaktion mit einer Reaktionsgenerierung nach der Valence-Bond-Emulation nicht aufgerufen, kann es zu unbeabsichtigten Effekten kommen. Da der für
die Erkennung der Identität von Produkten verwendete Hashc
Mit der Funktion valence kann überprüft werden, ob ein erzeugtes Produkt eine korrekte Struktur besitzt. mode gibt dabei den Modus an, mit dem die Prüfung durchgeführt wird. Es stehen zur Auswahl: organic,
inorganic, ms, ms-inorganic, none, ... // emulation void change_bond_order(int ia1, int ia2, int delta, int single_aro_split=1); void change_free_els(int ia, int i); void conjugate_elsys(); bool valence(int mode=0);
# emulation change_bond_order $ia1 $ia2 $delta $single_aro_split change_free_els $ia $i conjugate_elsys set flag [valence $mode]Die Funktion
change_bond_order verwendet für das Brechen von Bindungen intern einige Heuristiken zur automatischen Verteilung der Elektronen auf die beiden neuen Elektronensysteme. Eine Bindung gilt dann als gebrochen, wenn beide Elektronensysteme
nach dem Bruch radikalisch sind. Dies bedeutet, daß beim Bruch von nicht cyclischen -Systemen die mesomere Grenzstruktur genommen wird, bei der an der zu brechenden Stelle nach Möglichkeit keine Doppel- oder Dreifachbindung
ist. Die Mesomerie weicht also einem Bindungsbruch aus." Gebrochene cyclische -Systeme zählen dann als Bindungsbruch, wenn single_aro_split gleich 1 bzw. nicht angegebenen ist. Welche der -Bindungen
einer Dreifachbindung gespalten wird, ist zufällig. Die eingebauten Heuristiken haben nur eine begrenzte Gültigkeit, für die die Emulationsfunktionen angewendet werden können. Mit der Beschränkung auf neutrale organische Verbindungen
und Oniumionen aus Hauptgruppenelementen, bei denen keine Oktettaufweitung stattgefunden hat, läßt sich dennoch fast die ganze organische Chemie formulieren. Eine Schwierigkeit ergibt sich bei Strukturen, die ein Stickstoffatom mit zwei -
und zwei -Systemen enthalten. Hier wird angenommen, daß dieses Atom zwei Elektronen zum betrachteten -System beiträgt, wobei es auch eines sein kann, was aber nicht eindeutig entschieden werden kann.
Es könnte sich um ein konjugiertes freies Elektronenpaar, eine Einfachbindung sowie eine Doppelbindung handeln oder um eine Kombination aus einer Dreifach- und einer Einfachbindung bzw. zweier Doppelbindungen, wobei das Stickstoffatom eine positive Formalladung
tragen würde. Formalladungen sind nicht Bestandteil der MO-orientierten Molekülstruktur, da sie mit der Delokalisation der Elektronen auch delokalisiert werden können. Die Besetzungszahl des -Systems kann nicht verwendet
werden, da man ja gerade dabei ist, die Zahl der Elektronen zu bestimmen, die eigentlich in diesem Elektronensystem enthalten sein sollten, und zudem könnten ja noch weitere solcher Atome an dem -System beteiligt sein. Spätestens
dann ist eine Unterscheidung nicht mehr möglich. Sind in Molekülen -Bindungen mit mehr als zwei Zentren oder Komplexbindungen in den Molekülen enthalten, kann die Valence-Bond-Emulation nicht verwendet werden. In solchen
Fällen muß auf die MO-orientierte Reaktionsgenerierung zurückgegriffen werden (siehe unten).-Systems im Benzol ein Elektronensystem mit sechs Elektronen. Jede -Bindung und jedes Lonepair sind auch jeweils ein Elektronensystem mit zwei Elektronen. Neben den einfach- (Radikale),
zweifach- und mehrfachbesetzten Elektronensystemen gibt es auch unbesetzte, wie im Fall eines nicht konjugierten Carbokations.Drei Funktionen sind für die Reaktionsgenerierung zuständig:
split_elsy)
combine_elsys)
change_electrons)
Um zwei Elektronensysteme zu verbinden übergibt man der Funktion combine_elsys die beiden Indizes der Elektronensysteme und den Typ des Elektronensystems, das gebildet werden soll. Als Typen gibt es folgende Möglichkeiten:
es_sigma
es_pi
es_default
Bei den ersten drei Möglichkeiten wird ein entsprechendes Elektronensystem gebildet, wobei auch Lonepairs (besetzt oder unbesetzt) den Typ es_pi besitzen.
Für es_sigma sollte man zwei -Systeme angeben, die nur ein einziges Atom enthalten. In diesem Fall ist klar, zwischen welchen Atomen das -System gebildet werden soll. Anderenfalls wird intern
versucht herauszufinden, wo die Bindung erzeugt werden soll, es kann aber nicht garantiert werden, daß diese Stelle auch der entspricht, die beabsichtigt war. Mehrzentren--Systeme können schrittweise aus besetzten bzw. unbesetzten
Lonepairs (ein Atom--Systemen) aufgebaut werden. Zunächst entsteht ein normales 2-Elektronen-2-Zentren--System, das dann nach und nach erweitert werden kann. Dazu verknüpft man das -System
mit einem unbesetzten Lonepair und erhält ein Dreizentren--System. Hierbei gibt es wiederum zwei Formen: -Systeme ohne Atom im Zentrum, wie die BBB-Bindung in Boranen, und das -System
mit Atom im Zentrum, wie die BHB-Bindung in Diboran, bei der das Wasserstoffatom das Zentrum bildet. Primär entsteht beim schrittweisen Aufbau eines Mehrzentren--Systems immer ein -System ohne Atom
im Zentrum. Soll ein Atom als Zentrum gesetzt werden, kann dies anschließend mit der setzbaren Eigenschaft EL_CENTER geschehen, die den Index des Atoms angibt, das zum Zentrum werden soll.
es_complex erzeugt eine Komplexbindung, die analog der Bildung von Mehrzentren--Systemen schrittweise aus einem besetzten -System und mehreren unbesetzten Orbitalen (-Systemen
an einem Atom ohne Elektronen) des Metallatoms aufgebaut und gegebenenfalls auch wieder in diese gespalten werden.
es_default erzeugt ein -System, wenn die zwei Atome kein Elektronensystem verbindet - ansonsten ein -System. Der Rückgabewert der Funktion ist der Index des gebildeten Elektronensystems.
Um die Indizes der Elektronensysteme zu erhalten, fragt man die Atomeigenschaft A_ELECSYSS bzw. die Nachbarschaftseigenschaft N_ELECSYSS ab und erhält die Indizes der Elektronensysteme an einem Atom bzw. die Elektronensysteme,
an denen die zwei angegebenen Atome beteiligt sind.
// Reaktionsgenerator int combine_elsys(int iel1, int iel2, RX_ELSY_TYPE typ=es_default); bool split_elsy(int ie, int ia1, int ia2, int &np1, int &np2, int iel1=-1); bool change_electrons(int ie, int delta);
# Reaktionsgenerator set new_els [combine_elsys $iel1 $iel2 $created_els_type] set flag [split_elsy $ie $ia1 $ia2 np1 np2 $iel1] set flag [change_electrons $ie $delta]
split_elsy spaltet das Elektronensystem mit dem Index ie zwischen den Atomen ia1 und ia2. iel1 gibt die Zahl der Elektronen an, die das Elektronensystem erhalten soll, zu dem das Atom ia1
gehört. Ist die Zahl negativ, werden die Elektronen nach Möglichkeit so verteilt, daß in den neuen Elektronensystemen so viele Elektronen enthalten sind, wie deren beteiligte Atome zum alten Elektronensystem beigesteuert haben, was allerdings
dieselben Beschränkungen wie die automatische Verteilung der Elektronen bei der Spaltung von -Bindungen nach der Valence-Bond-Methode (siehe oben) hat. In den Variablen np1 und np2 stehen nach dem Aufruf
die Indizes der neu gebildeten Elektronensysteme, wobei einer der beiden auch dem Index des alten Elektronensystems entsprechen kann. Der Rückgabewert ist ein Flag, das angibt, ob das Elektronensystem gespalten werden konnte (C++: true / Tcl:
1 erfolgreich bzw. false / 0 nicht).
Handelt es sich bei dem zu spaltenden Elektronensystem um ein -System mit mehr als zwei Atomen, wird das Atom ia1 aus dem -System herausgenommen und es entsteht am Atom ia1 ein
-System mit einem Atom und iel1 Elektronen. Ist iel1 negativ oder nicht angegeben, erhält das -System null Elektronen. Das Atom ia2 hat in diesem Fall keine Bedeutung
und muß auf 0 gesetzt werden. np1 enthält nach dem Aufruf die Nummer des gebildeten -Systems. Analoges gilt für Komplexbindungen.
Schließlich kann man mit change_electrons die Zahl der Elektronen des Elektronensystems ie bei positivem delta um delta erhöhen bzw. bei negativem um delta erniedrigen.
new_atom
erhält mit ipernr die Ordnungszahl des zu erzeugenden Atoms und gibt den Index dieses hinzugefügten Atoms zurück. Im Vektor elss stehen die Indizes der Elektronensysteme, die am neuen Atom hängen. In diesem Fall
gibt man als Reaktivität die Geschwindigkeitskonstante pseudoerster Ordnung k1 an (k2: Geschwindigkeitskonstante zweiter Ordnung):Bei komplexen Molekülen besteht auch die Möglichkeit das Molekül vom File einzulesen (siehe unten).
In manchen Situationen ist man auch an den Coprodukten nicht interessiert. Dies kann beispielsweise in der Massenspektroskopie für die abgespaltenen Neutralfragmente oder das entstehende Wasser bei einer Esterbildung in der kombinatorischen Chemie der
Fall sein. Diese Aggregate können mit delete_aggregate gelöscht werden, wobei agr_id den Index des Aggregates angibt, das in den beiden erwähnten Beispielen aus einem einzigen Molekül besteht. Diesen Index erhält
man unter anderem von der Atomeigenschaft A_AGGREGATE.
int new_atom(int ipernr, vector<int> &elss); bool delete_aggregate(int agr_id);
set ia [new_atom $ipernr elss] set flag [delete_aggregate $agr_id]
h-bridge, ring, catenan, etc.. Desweiteren kennt jede Gruppe noch ihre Zugehörigkeit zu einem Aggregat, Molekül, Atom oder Elektronensystem
(chemtyp) und zwei Flags, make_aggr und splittable. make_aggr = 1 gibt an, daß die Gruppe zwei oder mehrere Moleküle als Aggregat zusammenhält, wenn die Atome/Elektronensysteme aus verschiedenen Molekülen
stammen. Ist make_aggr = 0 wird die Gruppe gelöscht bzw. gespalten, wenn nach der Reaktion nicht alle Atome/Elektronensysteme zu einem Molekül gehören. Ob die Gruppe in diesem Fall gelöscht wird oder nur gespalten wird, liegt
am Flag splittable. Ist dieses Flag 1, wird gespalten und die neuen Gruppen erhalten die Werte der gespaltenen Gruppe; ist es 0, wird die Gruppe gelöscht. Die übrigen Funktionen erklären sich von selbst.// Gruppen Steuerung int new_group(const char *name, const char* chemtyp, int idx,
int make_aggr=1, int splittable=0); int new_group(const char *name, ds_chem_type chemtyp, int idx,
int make_aggr=1, int splittable=0); bool delete_group(int gr_id); bool add_atom_to_group(int gr_id, int at_id); bool delete_atom_from_group(int gr_id, int at_id); bool add_elsy_to_group(int gr_id, int el_id); bool delete_elsy_from_group(int gr_id, int el_id); void get_all_groups_of_type(const char *name, vector<int> &nums); void get_groups_of(const char* chemtyp, int idx, vector<nt> &nums); const char* get_type_of_group(int grp_idx); const char* group_belongs_to(int grp_idx); ds_chem_type group_belongs_to_ctyp(int grp_idx);
// Gruppen Steuerung set idx [new_group $name $chemtype $idx $make_aggr $splittable] set flag [delete_group $gr_id] set flag [add_atom_to_group $gr_id $at_id] set flag [delete_atom_from_group $gr_id $at_id] set flag [add_elsy_to_group $gr_id $el_id] set flag [delete_elsy_from_group $gr_id $el_id] get_all_groups_of_type $name grp_idxs get_groups_of $chemtype $dx set name [get_type_of_group $grp_idx] set tpye [group_belongs_to $grp_idx]
new_prop werden vier Parameter übergeben, der Name der neuen Eigenschaft prop_name, der Datentyp der Eigenschaft (int, double, vector<int>,
vector<double>, e_bit64 oder char*), der Chemietyp (atom, electronsystem, aggregate, molecule oder group) sowie ein Flag us4hash, ob diese Eigenschaft für die Berechnung der Hashcodes verwendet
werden soll (1 = ja, 0 = nein). Diese so erzeugten Eigenschaften können dann während der Reaktion gesetzt werden (siehe unten).
// create new property
bool new_prop(const char* prop_name, const char* dat_type, \
const char* chm_type, int us4hash=0);
bool new_prop(const char* prop_name, const char* dat_type, \
const char* chm_type, rx_prent& ent, int us4hash=0);
bool new_prop(const char* prop_name, proptype dat_type, \
ds_chem_type chm_type, rx_prent& ent, int us4hash=0);
set flag [new_prop $prop_name $dat_type $chm_type $us4hash]Die Namen der Eigenschaften sollten der Konvention entsprechen, daß Eigenschaften für Atome mit
A_, für Elektronensysteme mit EL_, für Gruppen mit G_, für Nachbarn mit N_, für
Moleküle mit M_ und für Aggregate mit AG_ beginnen. Eigenschaften für das Ensemble (E_) können zwar erzeugt werden, werden allerdings nicht für Folgereaktionen gespeichert, da das Produktensemble
in EROS7 in die einzelnen Aggregate gespalten und als solche gespeichert wird. Dies dient der Reduktion des Speicherbedarfs. Für die Funktionen, in denen rx_prent& ent vorkommt, enthält ent nach dem Aufruf den richtigen
Entry, der zum Abfragen der Eigenschaft verwendet werden kann.EL_CENTER können während der Reaktion folgendermaßen gesetzt werden:
// set properties (ensemble vars)
bool set_prop(const int &var, const char* prop_name, int idx1=0, \
int idx2=0);
bool set_prop(const double &var, const char* prop_name, int idx1=0, \
int idx2=0);
bool set_prop(const vector<int> &var, const char* prop_name, int idx1=0, \
int idx2=0);
bool set_prop(const vector<double> &var, const char* prop_name, int idx1=0, \
int idx2=0);
bool set_prop(const e_bit64 &var, const char* prop_name, int idx1=0, \
int idx2=0);
bool set_prop(const char *const &var, const char* prop_name, int idx1=0, \
int idx2=0);
//
bool set_prop(const int &var, const rx_prent &p, int idx1=0, \
int idx2=0);
bool set_prop(const double &var, const rx_prent &p, int idx1=0, \
int idx2=0);
bool set_prop(const vector<int> &var, const rx_prent &p, int idx1=0, \
int idx2=0);
bool set_prop(const vector<double> &var, const rx_prent &p, int idx1=0, \
int idx2=0);
bool set_prop(const e_bit64 &var, const rx_prent &p, int idx1=0, \
int idx2=0);
bool set_prop(const char *const &var, const rx_prent &p, int idx1=0, \
int idx2=0);
set flag [set_prop var $prop_name $idx1 $idx2]In den genealogischen Variablen kann man Angaben wie die Anregungsenergie des Aggregates oder Umlagerungszähler speichern. Außerdem kann man mit ihrer Hilfe steuern, welche Reaktionstypen für Folgereaktionen zugelassen sind.
save_active_ens_as_product zum Speichern der Reaktion und set_active_ens(1), um erneut mit dem Edukt zu beginnen, verwendet
werden. Die Ensembles werden dabei in der Reihenfolge, in der sie erzeugt werden, durchnumeriert. Die Nummer des aktiven Ensembles kann mit der Funktion get_active_ens abgefragt werden. Wird keine der Funktionen save_active_ens_as_product
oder save_ens_as_product aufgerufen, ist automatisch das letzte erzeugte Ensemble das Produkt der Reaktion.// ensemblesteuerung bool set_active_ens(int ie); int get_active_ens(); void save_active_ens_as_product(); bool save_ens_as_product(int ie); bool append_ens(int i); void empty_ens();
set flag [set_active_ens $i] set i [get_active_ens] save_active_ens_as_product set flag [save_ens_as_product $i] set flag [append_ens $i] empty_ens
Zwei weitere Gründe für die speziellen IO-Routinen sind:
Als Formate sind momentan CTX, SCREEN und CTX_MO implementiert.
Wenn mit der print_active_ens-Funktion gearbeitet wird, die const char* file_name als Übergabeparameter hat, oder man mit Tcl arbeitet, kann mit open_stream und close_stream das File zum Lesen und Schreiben
öffnet bzw. geschlossen werden.
// IO void print_active_ens(ostream& fp, const char *format); void print_active_ens(ostream& fp, e_format format=SCREEN); void print_active_ens(const char* format, const char* file_name); void open_stream(const char* name); void close_stream(const char* name); bool read_ens(const char* file_name); bool read_ens(istream& fp);
print_active_ens $format $file_name open_stream $name close_stream $name read_ens $file_name
result_phase der Klasse rx_eifc auf die Nummer der Phase zu setzen, in die die Produkte gehen sollen. Dabei ist darauf zu achten, daß der Wert nicht außerhalb
der existierenden Phasen liegt. Desweiteren sollte die Produktphase innerhalb des aktuellen Reaktors liegen. Soll ein Kinetikmodell verwendet werden, muß sie das auch, da die Produkte bei Phasenübergängen und Phasentransferreaktionen zwischen
Reaktoren eine Konzentration von 0.0 erhalten. Die kinetischen Berechnungen wären also falsch. Dies liegt daran, daß jeder Reaktor seine eigene Kinetik besitzt.Gesetzt wird die Produktphase wie folgt:
eifc->result_phase = 2;
set result_phase 2Zu Beginn jeder Reaktion hat die Variable
result_phase den Wert der Eduktphase. Die Phase des ersten Aggregates, die diese Reaktion gerade erzeugt, kann man auch aus der Variablen phase, die von EROS7 an die Regeln übergeben wird,
erhalten.eifc->symmetry_factor *= 2;
set symmetry_factor [expr $symmetry_factor*2]
Überprüft man die gelieferten Flags, kann man diese Warnungen zusammen mit der Unterdrükkung dieser Reaktionen im Regelteil INIT_RULES folgendermaßen abstellen:
eifc->putco("no_dicard_on_error", 1);
global tvar set tvar 1 putco no_dicard_on_error tvarHat man nicht in allen Fällen die Fehlerflags getestet, kann man vor dem Verlassen von
FUNC mit bool flag=eifc->get_err_ocur(); überprüfen, ob ein Fehler eingetreten ist (true) und gegebenenfalls den
Regelfunktionsaufruf mit return BAD; beenden. Mit eifc->set_warnings(true); bzw. mit false wird das Fehlerflag zurückgesetzt.reactivity bekanntgegeben wurde.
Die Nummern der Atome, Elektronensysteme und Gruppen bleiben während der ganzen Reaktionsschritte gleich, damit man sein Reaktionszentrum auch in den Produkten wiederfindet. Die Nummern der Aggregate und Moleküle muß man sich dagegen nach jeder
Modifikation des Ensembles wieder holen z.B. über die Atome. Da in den Reaktionen beispielsweise durch die Kombination zweier Elektronensysteme zu einem Elektronensysteme vernichtet werden können, aber die Indizes der anderen Elektronensysteme gleich
bleiben, kann es zu Löchern in der Reihenfolge der Nummern der Elektronensysteme kommen. Anfänglich sind alle Elektronensysteme konsekutiv durchnumeriert, aber nach der Reaktion gibt es beispielsweise das Elektronensystem Nummer 5 nicht mehr. Um so
etwas feststellen zu können, wenn man es nicht bei den Reaktionsfunktionen mitprotokolliert, gibt es spezielle Eigenschaften: E_N_..., E_LAST_... und ._EXIST. Die Punkte stehen dabei für AGGREGATES,
MOLECULES, GROUPS, ELECSYSS oder ATOMS bzw. der einzelne Punkt entsprechend für AG, M, G, E oder A. E_N_... liefert die Anzahl
der ... im aktuellen Ensemble, wohingegen E_LAST_... die höchste verwendete Indexnummer für ... liefert. Der Unterschied liegt in den ungültigen Indexnummern, der durch die Löschung von Aggregaten auch bei Atomen usw. auftreten
kann. Will man nun feststellen, ob ein bestimmter Index noch gültig ist, kann man sich die immer gültige Eigenschaft ._EXIST für diesen Index holen. 1 bedeutet, daß der Index gültig ist, und 0,
daß der Teil des Ensembles nicht (mehr) existiert.
Es gibt noch eine Reihe weiterer Eigenschaften, die nicht mit den Aggregaten/Molekülen gespeichert werden, sondern vom Regelinterface generiert werden. Alle Eigenschaften, die Indizes zurückliefern, gehören dazu:
-System ist (1=ja, 0=nein)-Systeme mit mehr als 2 Atomen sowie Setzen des Typs des -Systems. Ist der gesetzte Wert (Atomindex) 0, ist / wird es ein -System
ohne Zentrum.PREP_ROOT aufruft:
int idx=1, idxs;
const char *pname, *ptype;
while ((pname = eifc->prop_name(idx))) {
ptype = eifc->prop_type(pname);
idxs = eifc->prop_idxs(pname);
print << "Eigenschaft " << pname << " hat den Typ ";
print << ptype << " und braucht " << idxs;
print << " Indizes" << endl;
idx++;
}
set idx 1
while {$idx} {
set pname [prop_name $idx]
if ($pname) {
set ptype [prop_type $pname]
set idxs [prop_idxs $pname]
print "Eigenschaft $pname hat den Typ "
print "$ptype und braucht $idxs Indizes\n"
incr idx
} else {
set idx 0
}
}
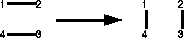
k = (2.5 + q(Edukt, Atom 2) - q(Produkt, Atom 3)) * 10-6:
int a1, a2, a3, a4; a1 = eifc->center(1); a2 = eifc->center(2); a3 = eifc->center(3); a4 = eifc->center(4); double qsige2, qsigp3; bool flag; // Eduktfunktion flag = eifc->prop(qsige2, "A_QSIG", a2); // Reaktionsgenerator eifc->change_bond_order(a1,a2,-1); eifc->change_bond_order(a3,a4,-1); eifc->change_bond_order(a2,a3,+1); eifc->change_bond_order(a1,a4,+1); // Produktfunktion flag = eifc->prop(qsigp3, "A_QSIG", a3); // Berechnung der Reaktivität // var_reactivity wurde unter INIT_RULES // mit put als reactivity übergeben var_reactivity = (2.5 + qsige2 - qsigp3) * 1.e-6; return OK;
set a1 [center 1] set a2 [center 2] set a3 [center 3] set a4 [center 4] # Eduktfunktion set flag [prop A_QSIG $a2 qsige2] # Reaktionsgenerator change_bond_order $a1 $a2 -1 change_bond_order $a3 $a4 -1 change_bond_order $a2 $a3 1 change_bond_order $a1 $a4 1 # Produktfunktion set flag [prop A_QSIG $a3 qsigp3] # Berechnung der Reaktivität # var_reactivity wurde unter INIT_RULES # mit put als reactivity übergeben set var_reactivity [expr (2.5 + $qsige2 - $qsigp3) * 1.e-6] return OK